This tutorial will walk you through how to use existing tracks on the UCSC Repeat Browser, as well as how to use it to view your own data. The Repeat Browser provides an easy way of visualizing genomic data on consensus versions of repeat families. This can be useful in a variety of ways; for instance if you’d like to study a particular transcription factor and its binding to transposable elements, the Repeat Browser can aggregate the data from every TE of the same class and display its binding on a consensus. The Repeat Browser is further described in Fernandes et al., 2020.
Using the Browser with Precomputed Data
The Repeat Browser functions in a manner analogous to the UCSC Genome Browser. Genomic data is displayed in a reference coordinate system. In the Repeat Browser “chromosomes” are consensus versions of repeats that are scattered throughout the human genome (roughly 55% of the genome is annotated by RepeatMasker as a repeat). We have taken existing genomic data already mapped to the human genome and “lifted” it to the Repeat Browser. Thus data from the (potentially) 1000’s of copies scattered around the genome all “pileup” on the consensus and can be viewed on the browser as individual mapping instances or coverage plots.
When you load the Repeat Browser, it will, by default, take you to the repeat L1HS.

You can type any repeat you know of in the search bar to move to that consensus. Alternatively you can click on the live links on this page. Let’s go the the repeat L1PA4. Zoom in to the 5’UTR by holding ctrl+mouse (or right click) to drag a zoom box or type L1PA4:1-1000 in the search box.

Once you are on the repeat you are interested in you can turn on and off tracks just like you would on the UCSC Genome Browser (by either using ctrl+mouse (or right click) or clicking on the track descriptions below the browser). Here we have turned on a few tracks, and displayed them in various display settings (dense, pack, full). We’ve also zoomed into the first 1000 bp of the element. Of note are the “meta-summits” tracks. These meta-summits suggest that the factor being displayed is binding most of the repeats of this type (all across the genome) at this location. You can verify this by looking at that factor’s individual subtrack (it will have nomenclature <first author_last author> and either be a “summit track” (individual genomic position mappings) or a coverage track (density coverage of each base by those mappings). Let’s verify the meta-summits by turning on those YY1 ChIP-SEQ coverage tracks from “Schmittges_Hughes 2016” from the “Coverage of Chip-Seq summits from large screens” track collection. Note that there is support for other meta-summits that could be shown on the meta-summits track. However these do not meet the score threshold (100) from the peak-caller output. You can access raw unfiltered peak files in the macs2 directory here.

Since many tracks on the Repeat Browser are composite tracks with LOTS of subtracks, displaying them all at once (especially in the full setting) can cause your browser to crash. Therefore we recommend using the meta peaks tracks to identify the coverage tracks you want to turn yourself. If you attempt to turn on the whole track from the browser window (instead of clicking on the track page and checking/unchecking boxes) you will only display a random subset of the data.
Note that you should always investigate how well the coverage track supports a meta peak before you get too excited about it.
You can click around the browser to see what else you can find. If you’d prefer to do more systematic analysis, download the tracks from the Table Browser or directly from our directories.
Let us know if you have any issues!
Using the Browser to Display your Own Data
Step 1: Process your data on the human genome assembly of your choice (hg19 or hg38).
The Repeat Browser is most commonly used to examine ChIP-SEQ data but potentially any coordinate data can be lifted. Indeed many standard annotations are already lifted and available as default tracks. While nothing stops you from lifting RNA-SEQ data, you might want to stop and think about if that’s what you really want to do (see FAQ).
For most ChIP-SEQ workflows you will map your reads to an assembly of the human genome. The two most recent assemblies are hg19 and hg38. Genomic mapping is typically done using a mapping algorithm like bowtie2 or bwa.
Since you are studying repeats you probably don’t want to get rid of multi-mapping reads (reads which map equally well to multiple parts of the genome)! Note that bowtie2 can be run in non-deterministic mode to assign multi-mapping reads randomly and test how random mapping decisions affect peak calling on both the human genome and the Repeat Browser.
After mapping, you will take your aligned data (typically in a bam or sam format) and call peaks with peak calling software like macs2. The result will be something like a bed file containing coordinates on the human genome that you now wish to view on the Repeat Browser. In most cases we are most interested in the summits of peaks which we can extend by an arbitrary number of nucleotides (typically +/- 5-50 bases) to smooth Repeat Browser peaks. We provide two samples files that you can use for this tutorial. These files are ChIP-SEQ summits from this highly recommended paper. ZNF765 is a KRAB Zinc Finger Protein which binds the transposable element families L1PA6, L1PA5 and L1PA4 in a quite characteristic way.
Sample Files:
ZNF765_Imbeault_hg19.bed [summits of hg19 mapping and peak calling; summits extended to 40 nt]
ZNF765_Imbeault_hg38.bed [the above file “lifted” to hg38]
Step 2: Lift from the human genome assembly (hg19 or hg38) to the Repeat Browser (hg38reps).
To lift you need to download the liftOver tool. “Lifting” is usually a process by which you can transform coordinates from one genome assembly to another. For the Repeat Browser we are “lifting” from the human genome to a library of consensus sequences.
You can download the appropriate binary from here:
https://hgdownload.soe.ucsc.edu/admin/exe/.
For instance, the tool for Mac OSX (x86, 64bit) is:
https://hgdownload.soe.ucsc.edu/admin/exe/macOSX.x86_64/liftOver
Once you have downloaded it you want to put in your path or working directory so that when you type “liftOver” into the command prompt you get a message about liftOver.
Once you have liftOver you need the liftOver file which provides mappings from the appropriate human genome assembly (hg19 or hg38) to the Repeat Browser (hg38reps).
You can download the files here:
hg19_to_hg38reps.over.chain [transforms hg19 coordinate to Repeat Browser coordinates]
hg38_to_hg38reps.over.chain [transforms hg38 coordinate to Repeat Browser coordinates]
Now you have all three ingredients to lift to the Repeat Browser:
1) Your hg38/hg19 data
2) Your hg38 or hg19 to hg38reps liftover file
3) The liftOver tool
You can use the following syntax to lift:
liftOver -multiple <hg38/hg19 data> <hg38/hg19 liftover file> <Repeat Browser output file> <unmapped file>
The “Repeat Browser file” is your data now in Repeat Browser coordinates. The unmapped file contains all the genomic data that wasn’t able to be lifted. This should mostly be data which is not on repeat elements. You don’t need this file for the Repeat Browser but it is nice to have. The “multiple” flag allows liftOver from the human genome to multiple Repeat Browser consensuses. This is important because hg38reps contains HERVK-full and HERVH-full (which are not part of normal RepeatMasker output) so data on HERVK-int annotations (on the genome) need to lift both to HERVK and HERVK-full (on the Repeat Browser). There are also a few cases where an interval of nucleotides (on the genome) is annotated as part of two repeats, so the multiple flag will allow proper lifting in those edge cases.
This procedure implemented on the demo file is:
liftOver -multiple ZNF765_Imbeault_hg38.bed hg19_to_hg38reps.over.chain ZNF765_Imbeault_hg38_hg38reps.bed ZNF765_Imbeault_hg38_hg38reps.unmapped
Now you have a file which can be visualized on the Repeat Browser! If you wish to turn it into a coverage track do the following (requires bedtools & the hg38reps.sizes “genome” file, and bedGraphToBigWig a UCSC tool available in the same download directory where you downloaded liftOver: https://hgdownload.soe.ucsc.edu/admin/exe/
bedSort ZNF765_Imbeault_hg38_hg38reps.bed ZNF765_Imbeault_hg38_hg38reps_sort.bed
followed by
bedtools genomecov -bg -split -i ZNF765_Imbeault_hg38_hg38reps_sort.bed -g hg38reps.sizes > ZNF765_Imbeault_hg19_hg38reps_sort.bg
followed by
bedGraphToBigWig ZNF765_Imbeault_hg19_hg38reps_sort.bg hg38reps.sizes ZNF765_Imbeault_hg19_hg38reps_sort.bw
Step 3: Visualizing on the Repeat Browser
Go to the Repeat Browser.
Click on My Data -> Custom Tracks
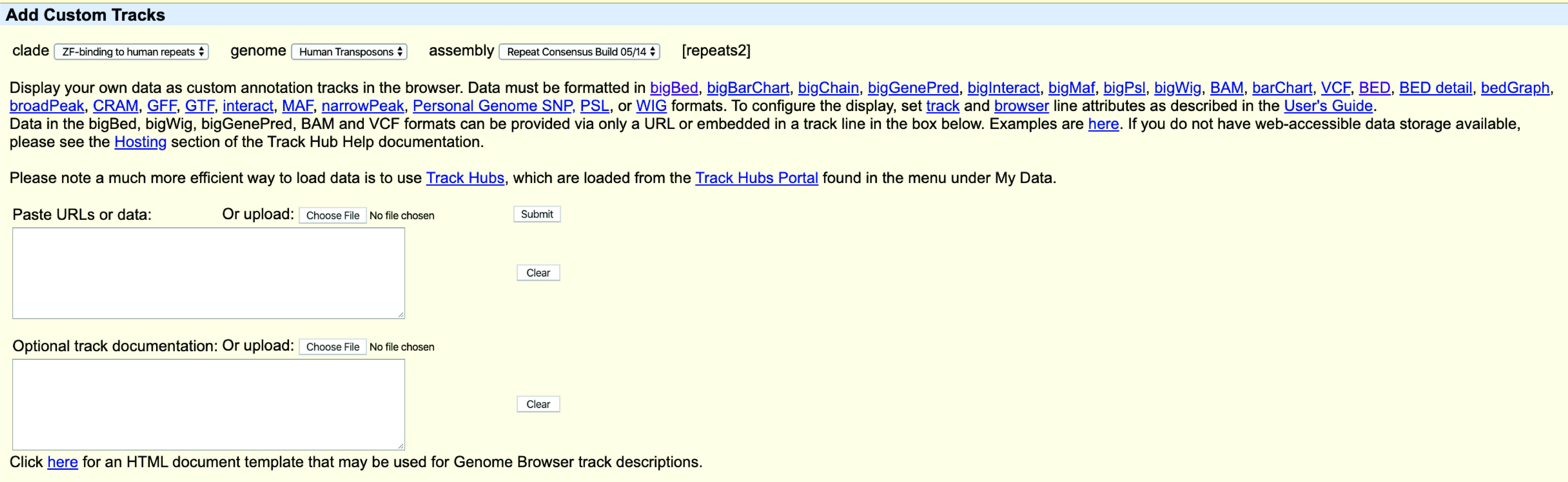
You can now upload the file (or copy and paste links to multiple files)
Click on Genome Browser
Your track will appear either as “User Track” (if no track information is in the file) or as a named track in the (Other) section.
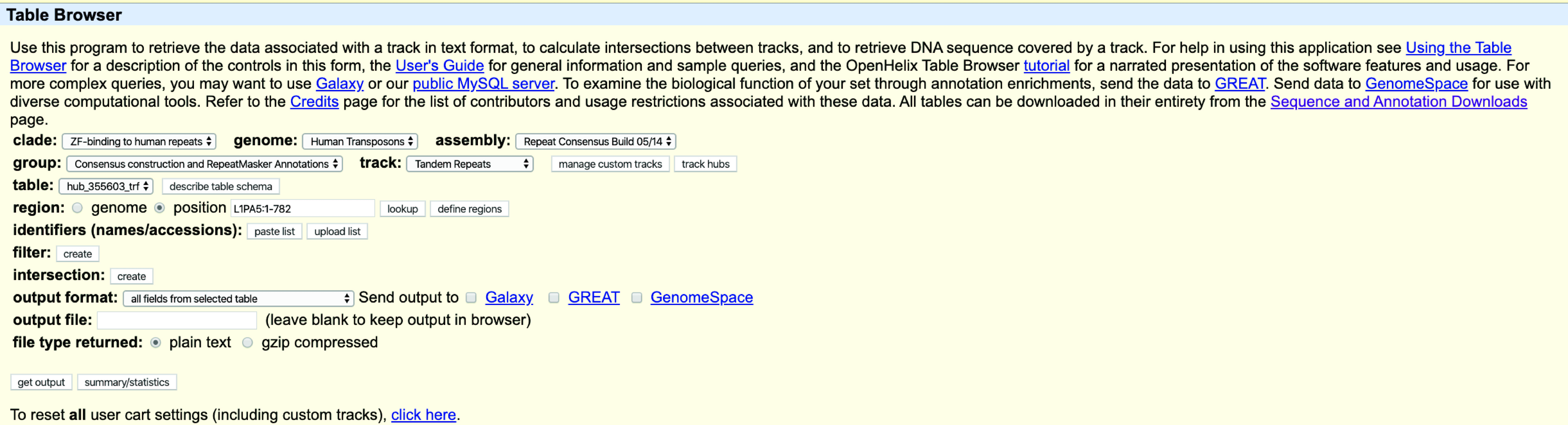
The sample file (hg19) should look as below on L1PA5: [click here for interactive session]
 You can go to any other repeat type by simply typing the name of the repeat into the search bar. A full list of all consensus repeats and their lengths is here
You can go to any other repeat type by simply typing the name of the repeat into the search bar. A full list of all consensus repeats and their lengths is here
Step 4: Analyze your data with existing tracks.
You can click on the Table Browser (Tools->Table Browser) to perform intersections, unions, etc through this user interface as you would normally with the Table Browser and the UCSC Genome Browser. You can also download tracks and perform this analysis on the command line with many of the UCSC tools.